
- +1 - (917) 400 89 00
- info@bbvreview.com
- UK , USA
Menu

Oct
- by Admin
- 0 Comments
Does hepatitis B virus have an Achilles heel?
Targeting the cccDNA episome offers hope of a future cure for HBV. Potential new compounds are outlined in this review but their effectiveness awaits to be demonstrated in on-going clinical studies
Chronic hepatitis B virus (HBV) infection can, over time, lead to the development of cirrhosis, hepatocellular carcinoma (HCC) and liver decompensation. With an estimated 350-400 million chronically infected individuals worldwide and 600,000 to 1 million HCC-related deaths annually, the infection constitutes a major global health problem (1,2). Antiviral treatment with an immunomodulator such as pegylated interferon (pegIFN) and nucleos(t)ide analogues (NUCs) has been the mainstay of therapeutic approaches to suppression of viral replication or clearance. NUCs act by inhibiting the viral reverse transcriptase/DNA polymerase and cause premature termination of DNA synthesis, both of which result in greatly reduced viral load levels. Loss of hepatitis B surface antigen (HBsAg), a most desirable outcome, is achievable in only a minority of patients (0-7% at best). Durable loss of HBeAg and seroconversion to anti-HBe in HBeAg positive patients ranges between 25- 33% and 12-22% following pegIFN or NUC treatment for one year respectively. In the case of HBeAg negative patients, loss of HBV-DNA is even lower at 15-20% with pegIFN, and although HBV-DNA negativity is high with NUCs after 1 year of treatment (51-93), relapse is quite frequent on stopping therapy (3).
Thus, unlike the case of IFN which is usually administered for a finite period of 1 year, NUC treatment is protracted in both HBeAg positive patients with no HBeAg seroconversion, and in HBeAg negative patients with detectable HBV DNA. NUC treatment is associated with an increasing risk of viral resistance through development of amino-acid substitutions in the reverse transcriptase (RT)/polymerase region of the viral genome. This is dependent on the type of NUC, with Entecavir and Tenofovir having the best resistance profiles (1.8% and 0% over 5 years respectively), whilst cumulative resistance with Adefovir and Lamivudine stands at 29% and 70% respectively, after 5 years of treatment (4,5).
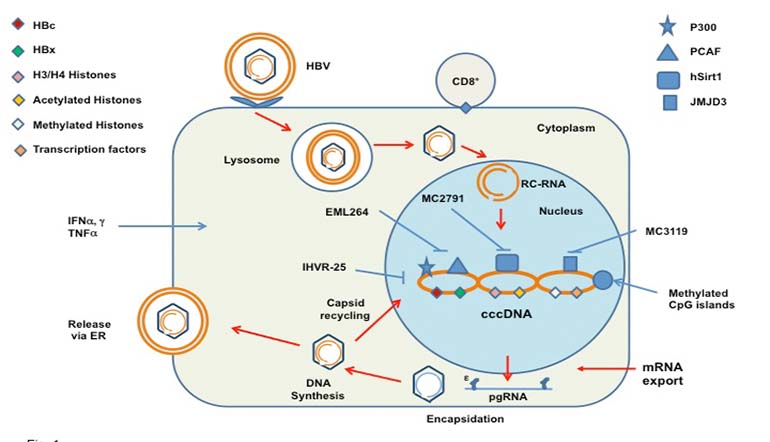
Premature withdrawal of antiviral treatment results in resumption of viral replication, made possible by the persistence of the episomal covalent closed circular (ccc) DNA. Following attachment to the hepatocyte membrane, endocytosis and uncoating, the viral genome in the form of relaxed circular DNA (RC-DNA) enters the nucleus where the partially double stranded DNA genome of the virus is converted through a number of steps into the cccDNA molecule (6,7). In this form, the cccDNA constitutes the template for transcript synthesis for production of the necessary viral proteins. One of these transcripts, the pregenomic (pg) RNA, is encapsidated within immature viral core particles where RC-DNA synthesis takes place. Firstly, the pgRNA is used as the template for the synthesis of the negative-sense DNA strand by reverse transcription, and this in turn forms the template for the synthesis of the partial positive-sense DNA strand. This process takes place in the maturing core particles in the cytoplasm, from whence most become fully formed virions following the acquisition of their outer HBsAg containing envelope (8) (Fig. 1). Some of the core particles though are recycled to the nucleus to replenish and maintain the cccDNA pool of about 20-50 copies per hepatocyte in the case of HBeAg positive patients (6). The size of this pool ultimately determines the rate of viral replication and therefore the levels of HBV DNA in serum. It follows that HBeAg negative patients with detectable HBV DNA harbor in their hepatocytes smaller size pools of cccDNA. A reduction in the cccDNA pool may occur during cell division (9). It should also be borne in mind that cccDNA persists in hepatocytes even after seroconversion (anti-HBs) and may lead to reactivation of viral replication after immunosuppression for solid organ transplantation or for other conditions.
As current antiviral treatments do not appear to significantly affect the cccDNA pool (10), there is a need for additional cccDNA targeting compounds. The most effective means of eliminating cccDNA-harbouring hepatocytes is through cytolysis, which existing antivirals are incapable of achieving and more recent immunotherapeutic approaches have failed to have an impact on (11). cccDNA elimination can only be achieved through lysis of infected cells which may be targeted by cytotoxic CD8+ T and natural killer cells, as well as IFN-α, -γ and TNF-α (12,13).
For us to talk about an HBV cure, the cccDNA episome must be targeted effectively and either eliminated or its formation/function prevented. It therefore constitutes its Achille’s heel, but currently our quiver is devoid of arrows which may be able to deliver the fatal blow. Recently however, new approaches targeting this stage of the viral life cycle have come to light. Compound IHVR-25
developed by the Institute for Hepatitis and Virus Research, PA, is currently in the preclinical stage of development, but detailed data on efficacy is presently lacking. Degradation of cccDNA in the absence of hepatotoxicity has recently been reported by Lucifora et al (14). Interferon-α and lymphotoxin-β receptor activation up-regulated the APOBEC3A and APOBEC3B cytidine deaminases, respectively. Their action mediated by the presence of the core protein expressed in HBV-infected cells (primary hepatocytes, and human liver needle biopsies) led to cytidine deamination, apurinic/apyrimidinic site formation, and eventually cccDNA degradation (14).
Transcription using the cccDNA minichromosome is controlled by a number of epigenetic factors which are recruited onto the cccDNA such as histones H3 and H4, transcription factors (CREB, ATF, STAT1 and STAT2) and chromatin modifying enzymes (15,16). All of these in their own right constitute potential targets for antiviral treatment. In addition, the viral core and X proteins also form part of the cccDNA/epigenetic factor complex. Cellular histone acetyltransferases and deacetylases recruited onto the cccDNA modulate histone acetylation which in turn controls cccDNA transcription. Thus, compounds which can inhibit the function/binding of these factors to the cccDNA constitute possible antivirals against HBV, provided they do not cause cellular toxicity. Evidence that such compounds hold promise has been obtained using the HepG2 and HepAD38 cell lines and small molecules/drugs that affect cccDNA chromatin bound modifying enzymes. It has been shown that such drugs modulate viral transcript synthesis and therefore replication capacity. Combined inhibition of p300 and PCAF histone acetyltransferases (compound-EML264) or stimulation of hSirt1/2 histone deacetylase activity (compound MC2791) had a negative effect on HBV replication mirrored by a concurrent reduction in pgRNA. A similar effect was obtained following inhibition of JMJD3 histone demethylase with compound MC3119 (17). It is also known that the level of CpG island methylation of the cccDNA is correlated with impaired HBV productivity in vivo (18). Therefore, cccDNA methylation status may also be an important mechanism of viral transcriptional control worth investigating further.
Although some progress has been made in targeting the cccDNA minichromosome, more needs to be done by way of small molecule discovery. We must await the results of clinical trials, if any of these compounds reach that stage. If they prove efficacious, such molecules may in future be used either alone, or more likely, in combination with existing or new therapies, in the quest of an HBV cure.
Figure 1 – A simplified diagram of the HBV life cycle showing hepatocyte attachement, endocytosis, uncoating and delivery of RC-DNA to the nucleus. Encapsidation of pgRNA is followed by reverse transcption and DNA synthesis, budding of maturing nucleocapsids through the endoplasmic reticulum membrane (ER) with acquisition of their outer envelope of HBsAg and exocytosis. Also shown are the cccDNA minichromosome and associated epigenetic and viral factors. Potential inhibitors of epigenetic factors and known mediators of hepatocyte lysis such as CD8+ cytotoxic T cells and antiviral cytokines are also shown. HBc and HBx= hepatitis B core and x proteins; PCAF=P300/CBP-associated factor; see text also.
Authors:
Peter Karayiannis BSc, PhD, FIBMS, FRCPath
Affiliation:
University of Nicosia Medical School, Cyprus
References:
1. Lee WM. Hepatitis B virus infection. N Engl J Med 1997;337:1733-45.
2. Ganem D, Prince AM. Hepatitis B virus infection-natural history and clinical consequences. N Engl J Med 2004;350:1118-29.
3. EASL clinical practice guidelines: management of chronic hepatitis B virus infection. J Hepatol 2012;57:167-185.
4. Viganò M, Lampertico P. Antiviral drugs for HBV liver disease. Expert Opin Biol Ther 2011;11:285-300.
5. Zoulim F. Hepatitis B virus resistance to antiviral drugs: where are we going? Liver Intl 2011;31 Suppl 1:111-6.
6. Tuttleman JS, Pourcel C, Summers J. Formation of the pool of covalently closed circular viral DNA in hepadnavirus-infected cells. Cell 1986;47:451-60.
7. Newbold JE, Xin H, Tencza M, Sherman G, Dean J, Bowden S, Locarnini S. The covalently closed duplex form of the hepadnavirus genome exists in situ as a heterogeneous population of viral minichromosomes. J Virol 1995;69:3350-7.
8. Nassal M. Hepatitis B viruses: Reverse transcription a different way. Virus Res 2008;134:235-49.
9. Summers J, Jilbert AR, Yang W, Aldrich CE, Saputelli J, Litwin S, Toll E, Mason WS. Hepatocyte turnover during resolution of a transient hepadnaviral infection. Proc Natl Acad Sci U S A 2003;100:11652-9.
10. Manesis EK, Papatheodoridis GV, Tiniakos DG, Hadziyannis ES, Agelopoulou OP, Syminelaki T, Papaioannou C, Nastos T, Karayiannis P. Hepatitis B surface antigen: relation to hepatitis B replication parameters in HBeAg-negative chronic hepatitis B. J Hepatol 2011;55:61-8.
11. Baltayiannis G, Karayiannis P. Treatment options beyond IFNα and NUCs for chronic HBV infection: expectations for tomorrow. J Viral Hepat 2014;21:753-61.
12. Guidotti LG, Rochford R, Chung J, Shapiro M, Purcell R, Chisari FV. Viral clearance without destruction of infected cells during acute HBV infection. Science 1999;284:825-9.
13. Guidotti LG, Ishikawa T, Hobbs MV, Matzke B, Schreiber R, Chisari FV. Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes. Immunity 1996;4:25-36.
14. Lucifora J, Xia Y, Reisinger F, Zhang K, Stadler D, et al. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science 2014;343:1221-8.
15. Pollicino T, Belloni L, Raffa G, et al. Hepatitis B virus replication is regulated by the acetylation status of hepatitis B virus cccDNA-bound H3 and H4 histones. Gastroenterology 2006;130:823-37.
16. Belloni L, Pollicino T, Cimino L, Raffa G, Raimondo G, Levrero M. Nuclear HBx binds in vivo on the HBV minichromosome, modifies the epigenetic regulation of ccc-DNA function and potentiates HBV replication J Hepatol 2008;48:s25-s25.
17. Palumbo TG, Belloni L, Pediconi N, Levrero M. Targeting the cccDNA by epigenetic drugs inhibits HBV transcription and replication. Hepatology 2013;58 (issue S1):650A.
18. Guo Y, Li Y, Mu S, Zhang J, Yan Z. Evidence that methylation of hepatitis B virus covalently closed circular DNA in liver tissues of patients with chronic hepatitis B modulates HBV replication. J Med Virol 2009;81:1177-83.

